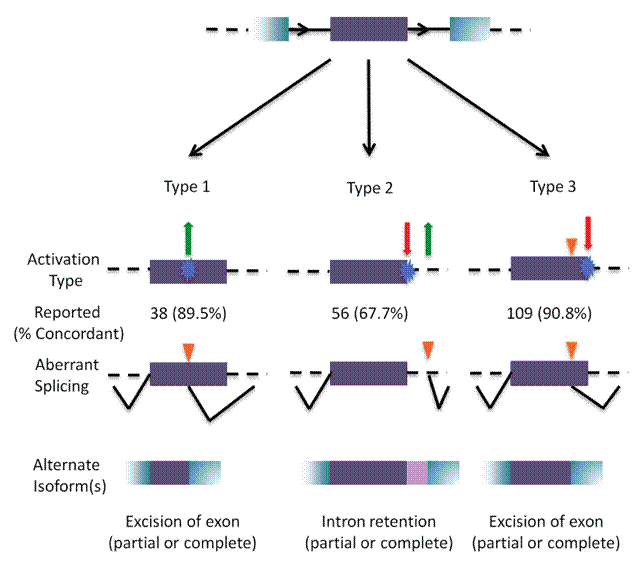
Recently, we published 2 papers describing our unifying framework for non-coding mutation analysis (Mucaki et al. BMC Medical Genomic, 2016; http://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-016-0178-5, and Caminsky et al. Human Mutation, 2016; http://onlinelibrary.wiley.com/doi/10.1002/humu.22972/full). Among the results were SNP analyses of transcription factor binding site mutations. These gene regions are very rich in variation, but only a small percentage of variants significantly alter the strengths of transcription factor binding sites. Knowing which sites are affected is important for mutation detection in these regions. The information theory-based models on which these SNP interpretations were based were obtained using a new approach just published in Nucleic Acids Research:
(Lu et al. 2016;http://nar.oxfordjournals.org/cgi/content/full/gkw1036?ijkey=l5dl5yGjigBzQqf&keytype=ref
I am still scratching my head about the current controversy regarding interpretation of VUS in breast cancer and other genetic diseases. I think the current focus on database discrepancies or differences in coding interpretation between commercial providers misses the key point. The pathogenic mutation yields in most exon-based sequencing studies alone are really quite poor. The amount and scope of non-coding variation completely dwarfs what is seen in coding regions. It is a more likely explanation for significant amount of the missing heritability in inherited predisposition and congenital disease than the discrepancies in coding sequences.
I am not claiming that the variants we prioritize with our framework are definitively pathogenic, but do believe that strategies that are narrowly focused on the genetic code itself won’t advance the field or help patients much. Clinical molecular geneticists seriously consider sequencing beyond coding regions and trying to interpret the variants detected in the regions. The incremental costs to do this aren’t exorbitant, and the excuse of ignorance about the meaning of such variants is simply not valid any longer.
Many non-coding mutations have been proven ‘anecdotally’; studies have not been designed to determine the incidence of these types of mutations, in part due to the higher densities of variants in non-coding regions, identifying the clinically relevant ones is more daunting. This has been compounded by the lack of bioinformatic and genomic methods to generate a reliable and comprehensive and high throughput validation of variants outside of coding regions with adverse functional consequences . Suffice it to say, there are many individual reports in the published literature, but they are not generally being systemically uncovered because of the narrow focus on changes in coding regions that affect amino acid sequences.
The problem is not only where the variants reside, but an overly conservative philosophy that fails to consider other interpretations for the effects of variants, even within coding regions. It’s not just non-coding regions that contain missing pathogenic variants, but also coding variants where the change in the amino acid code may not be the source of the disease pathology. There are actually numerous examples of this phenomenon (and a number of good reviews eg. Cartegni et al (https://www.ncbi.nlm.nih.gov/pubmed/11967553), however most genetic testing labs (commercial or academic) do not look for them proactively. This is the problem of overreliance on databases. If the authors of a paper describing a mutation are solely focused on changes in the amino acid code (most are), the cited reference will miss this
This is an example of a breast cancer predisposing mutation that affects mRNA processing (ie. exon skipping) even though it produces a premature termination of translation or stop codon: Peterlongo et al. 2014 (
http://hmg.oxfordjournals.org/content/24/18/5345.short). You can appreciate that if the exon containing the stop codon is spliced out prior to translation, then that particular stop codon is not activated.
Another example is this rare mutation causing
multiple Acyl-CoA dehydrogenation deficiency (Olsen et al. 2014; https://www.ncbi.nlm.nih.gov/pubmed/24123825?dopt=Abstract).. While the change appears to result in a missense mutation, it simultaneously introduces multiple RNA binding protein binding sequences for proteins that suppress exon recognition and weakens overlapping binding sequences that enhance recognition of the same exon. The result is that the exon is skipped during mRNA splicing, and the missense change is never introduced into the protein because the exon skipping event alters the reading frame of the mRNA.
In our recent review article (
https://f1000research.com/articles/3-282/v2), we compile 203 published examples of cryptic splicing mutations involving many different disorders analyzed by information theory with experimental validation. Some of the activated cryptic splice sites are exonic and others are non-coding, ie. intronic.
There is inevitably some bias against the reporting of intronic cryptic splicing mutations, because these sequences are not routinely determined in either research or clinical studies. Besides these classes, our studies also identify variants that alter transcription factor binding site strength and mRNA stability (in untranslated regions of mRNAs).
The exchange of mutation information about inherited breast cancer among various testing companies (except Myriad) has increased confidence in mutation interpretation. Those with rare mutations that are not shared among multiple patients do not benefit from this exchange. But these are generally based almost entirely on variants that cause amino acid substitutions or nonsense codons. I contend that such exercises, while very useful, are simply not scalable to the true volumes of all variants found in genes, and they ignore other mechanisms of pathogenicity such as those described above.
To reiterate, my argument is that current clinical molecular diagnostic practices will continue to leave many patients without known pathogenic mutations. Until this point of view changes and we seriously focus on functional and bioinformatic methods to analyze and prioritize VUSs thoughout genes, there will be a lot of frustration about the lack of results among the companies, academics and the patients they are purporting to help. We should also question whether the cost of testing can be justified, with the knowledge that a significant amount of genetic real estate is not being sequenced nor interpreted.
Peter K. Rogan