
Lu R and Rogan PK. Transcription factor binding site clusters identify target genes with similar tissue-wide expression and buffer against mutations [version 1 ]. F1000Research 2018, 7:1933 (DOI: 10.12688/f1000research.17363.1): https://f1000research.com/articles/7-1933/v1
December 7, 2018. New article describing database of validated mRNA splicing mutations published
Shirley BC, Mucaki EJ and Rogan PK. Pan-cancer repository of validated natural and cryptic mRNA splicing mutations [version 1; referees: awaiting peer review]. F1000Research 2018, 7:1908 (https://doi.org/10.12688/f1000research.17204.1
Website to perform database search: https://validsplicemut.cytognomix.com/
November 28, 2018. New automated cytogenetic biodosimetry article accepted for publication
RADIATION DOSE ESTIMATION BY COMPLETELY AUTOMATED INTERPRETATION OF THE
DICENTRIC CHROMOSOME ASSAY
Li, Yanxin; Shirley, Ben; Wilkins, Ruth; Norton, Farrah; Knoll, Joan; Rogan,Peter
Radiation Protection Dosimetry, in press
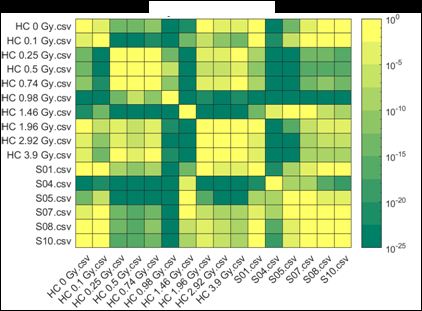
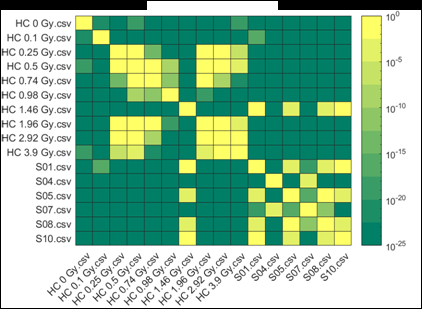
Figure 2 (from the article): Representative heat maps of chromosome object count distributions for Health Canada calibration (HC ##Gy.csv) and exercise (S##.csv) samples. Show: legend indicates significance of pairwise comparisons of object counts in (bottom panel) unselected vs. (top panel) Model B_C filter (5) selected images from each sample. Values close to 10^0 are not significant, whereas values such as 10^-20 are significant differences between samples. The legend displays probability thresholds of Wilcoxon signed rank tests of chromosome object counts between each pair of samples from the HC laboratory. Similar results were obtained for other chromosomal features.

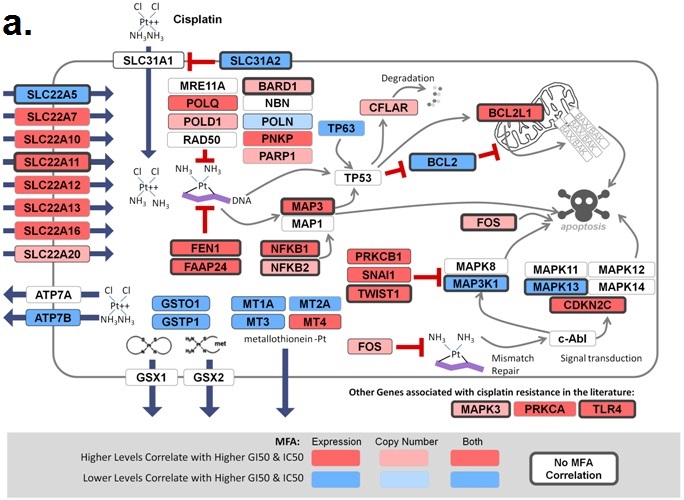
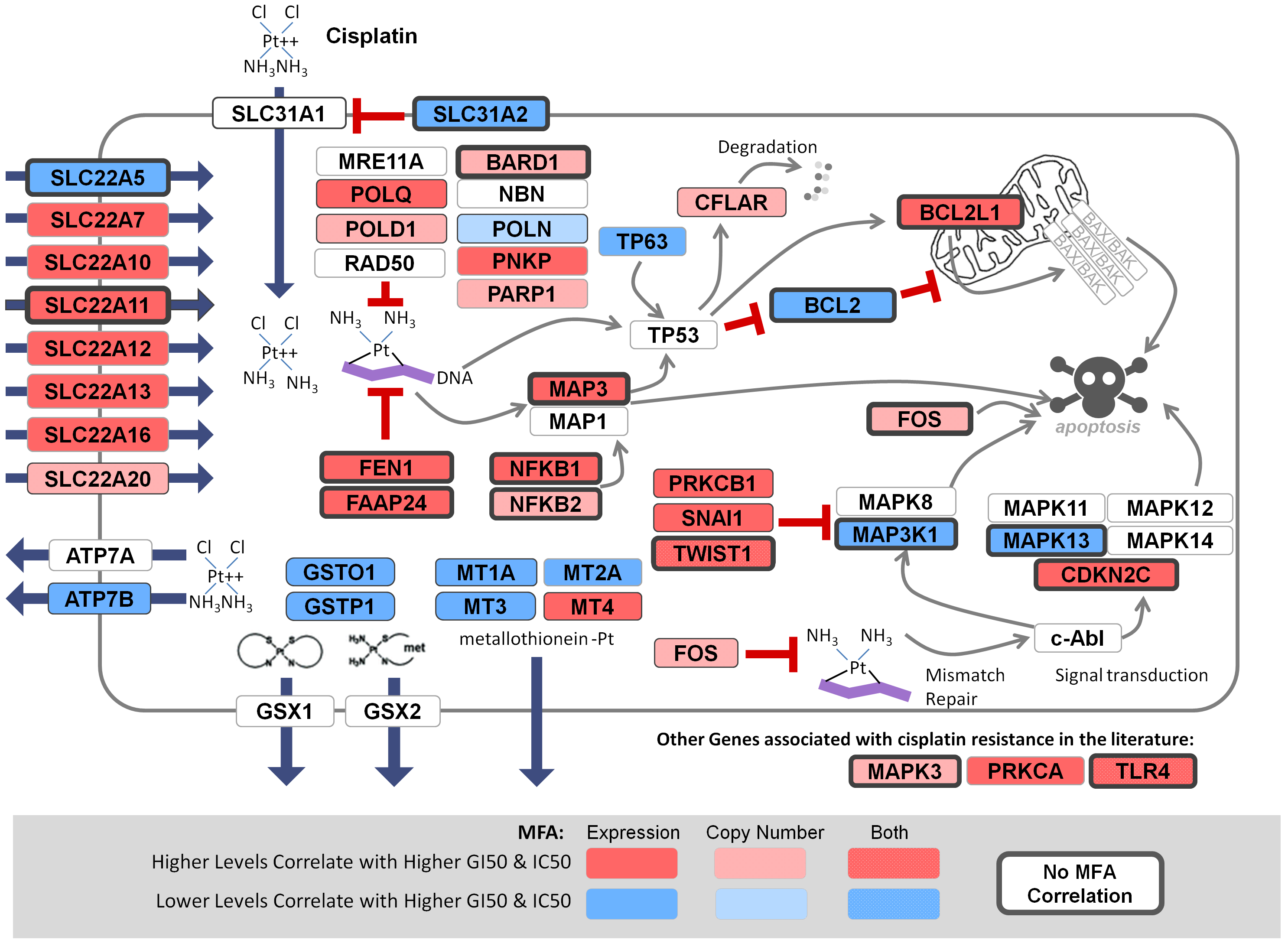
November 6, 2018. Article on prediction of platin chemotherapy response accepted for publication
Mucaki EJ, Zhao J, Lizotte D, Rogan PK. Predicting response to platin chemotherapy agents with biochemically-inspired machine learning. Signal Transduction and Targeted Therapy, in press.
A preprint of this article has been released on the BioRxiv server: https://doi.org/10.1101/231712
October 19, 2018. Presentations at the 7th Northeast Regional Chromosome Pairing Conference
CytoGnomix and the University of Western Ontario presented several papers about differential accessibility of single copy FISH probes to metaphase chromosomes at the Chromosome Pairing conference, held at Laurentian University, Sudbury, Ontario, Canada. We are grateful to Prof. Thomas Merritt for inviting us to participate in this exciting conference. The following talks were presented:
Seana Hill (Knoll and Rogan labs): Defining differentially accessible domains within human
metaphase homologues.
Jerry Wang (Knoll and Rogan labs): Expanding the catalog of differentially compacted loci
on homologous chromosomes in the human genome.
Peter Rogan (Rogan lab): A proposed mechanism to establish differential allelic
accessibility to homologous metaphase chromosomes.
Sept. 11, 2018. New publication about promoter gene variants in BRCA1 and BRCA2 that alter gene expression
In a collaboration with the ENIGMA Consortium members, we have used bioinformatic and functional genomic analysis to identify gene variants that affect the expression of the BRCA1 and BRCA2 genes. The article is:
Burke LJ, Sevcik J, Gambino G1, Tudini E, Mucaki EJ, Shirley BC, Whiley P, Parsons M, DeLeeneer K, Gutiérrez-Enríquez S, Pena MS, Caputo S, Santana dos Santos E, Soukupova J, Janatova M, Zemankova P, Lhotova K, Stolarova L, Borecka M, ENIGMA consortium, Edwards S, Blok R, Hansen TvO, Diez O, Vega A, Claes K, Rouleau E, , Radice P, Peterlongo P, Rogan PK, Caligo M, Spurdle AB and Brown MA. BRCA1 and BRCA2 5’ non-coding region variants identified in breast cancer patients alter promoter activity and protein binding, Human Mutation.
Link to full text of accepted paper: http://www.cytognomix.com/?attachment_id=4541
Permanent link: https://doi.org/10.1002/humu.23652
Aug. 13, 2018. Differential accessibility to homologous chromosomal loci confirmed by international consortium
A large international consortium based at Harvard University has demonstrated parental homolog-specific differences in chromatin accessibility on human chromosome 19:
Nir et al. BioRxiv doi: 10.1101/374058 (Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling)
This work reproduces previous reports previously published by CytoGnomix scientists using our patented scFISH™ probes:
Khan et al. Molecular Cytogenetics 2014 7:70 (Localized, non-random differences in chromatin accessibility between homologous metaphase chromosomes)
Khan et al. Molecular Cytogenetics 2015 8:65 (Reversing chromatin accessibility differences that distinguish homologous mitotic metaphase chromosomes)
June 13, 2018. Article in Fast Forward
The 2018 Impact report from the Southern Ontario Smart Computing for Innovation Platform (SOSCIP), which supports the development of a supercomputer version of the Automated Dicentric Chromosome and Dose Estimation (ADCI) system in IBM Blue Gene/Q, contains an article about our project:

https://www.soscip.org/wp-content/uploads/2017/08/soscip_impactreport2018_pages.pdf
June 7, 2018. Presentations at upcoming international conferences

Population scale biodosimetry with the Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) software system. (Platform) Rogan PK, Ali, S, Li Y, Shirley B, Wilkins R, Flegal F, Cooke R, Peerlaproulx T, Waller E, Knoll JHM. EPR Biodose 2018, June 11-15, Munich Germany
Optimization of image selection in Automated Dicentric Chromosome Analysis. Li Y, Shirley B, Wilkins R, Flegal, F, Knoll JHM, Rogan PK. EPR Biodose 2018, June 11-15, Munich Germany.
Predicting exposure to ionizing radiation by biochemically-inspired genomic machine learning. Rogan PK, Zhao JZL, and Mucaki EJ. EPR Biodose 2018, June 11-15, Munich Germany.

Comprehensive prediction of responses to chemotherapies by biochemically-inspired machine learning. (Best Poster session) Rogan PK, Zhao JZL, Mucaki EJ. European Society of Human Genetics 2018, June 16-19, Milan Italy.
May 29, 2018. Change to cytognomix.org URL
As of today, we have transitioned the website cytognomix.org to:
http://academic.cytognomix.com
The site contains our published articles, lectures and presentations about human genetics and molecular biology. All of the legacy content at this site (1980-2007) will be preserved on the new site.
Please update your browser bookmarks to reflect this change.
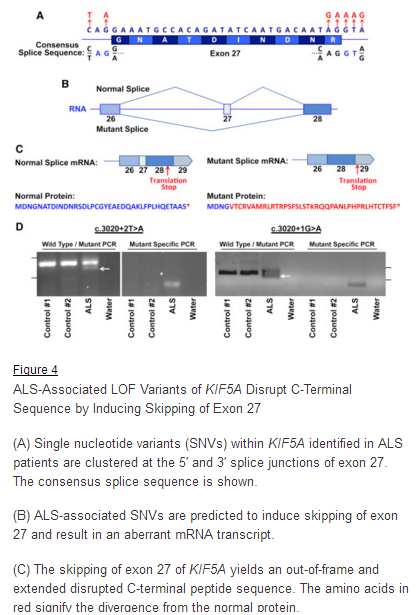
March 29, 2018. Mutation Forecaster: Key to discovery of mutations in novel ALS gene
CytoGnomix’s Mutation Forecaster: Key to discovery of mutations in novel ALS gene. Nicolas et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 97:1268-1283.e6, 2018
http://www.cell.com/neuron/fulltext/S0896-6273(18)30148-X
From our subscriber, Dr. John Landers (U. Mass. School of Medicine):
“We used the application ASSEDA (Automated Splice Site and Exon Definition Analyses) to predict any mutant mRNA splice isoforms resulting from these variants (Mucaki et al. 2013). This algorithm was chosen as it is known to have high performance in splice prediction (Caminsky et al., 2014). ASSEDA predicted a complete skipping of exon 27 for all variants, yielding a transcript with a frameshift at coding amino acid 998, the deletion of the normal C-terminal 34 amino acids of the cargo-binding domain, and the extension of an aberrant 39 amino acids to the C terminus (Table 3; Figures 4B and 4C). The presence of transcripts with skipped exon 27 was demonstrated by performing…”
April 9, 2018. Upcoming release of Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI)
We have just completed porting Windows ADCI from the MinGW C++ (32 bit) to Microsoft’s C++ (64 bit) compiled version. The release of this software in summer 2018 will contain this new version (v 2.0). ADCI now has access to 8 Gb of runtime memory, which should allow twice as many samples to be batch processed in a single run. We estimate at least 700 samples consisting of 500 images each can be analyzed in unattended operation.
Our primary motivation for building the 64 bit version was actually to write new code that exploits the onboard Nvidia graphics card (GPU) in the gaming computers that we recommend. Benchmarking of the Gradient Vector Flow code (that defines chromosome objects) indicates a speedup of ~20% when the main CPU is linked to the GPU. The speed of the Integrated Intensity Laplacian module (which measures chromosome crossectional width) will also benefit from the GPU link. GPU acceleration will be available later in the year.
Request a quote.
March 23, 2018. New preprint about target gene regulation by transcription factors
Clustered, information-dense transcription factor binding sites identify genes with similar tissue-wide expression profiles. BioRxiv, 2018.
doi: https://doi.org/10.1101/283267

March 13, 2018. Oral presentation on chemotherapy response in Best Poster session at ESHG 2018
On behalf of the Scientific Programme Committee of the European Conference of Human Genetics 2018 taking place in Milan, Italy from June 16 to June 19, 2018, we are pleased to inform you that the abstract entitled:
‘Comprehensive prediction of responses to chemotherapies by biochemically-inspired machine learning’
(Control No. 2018-A-2095-ESHG)
was among the best scored papers accepted for a poster presentation. Best Poster session takes place on Sunday, June 17, 2018 13:00 hrs, and consists of a 3 minute presentation followed by discussion at your electronic poster.
February 28, 2018. New publication on radiation biodosimetry based upon machine learning
We have published a new approach to devise gene signatures to detect radiation exposure (human, murine), and to quantify levels of exposure (murine):
Zhao JZL, Mucaki EJ and Rogan PK. Predicting ionizing radiation exposure using biochemically-inspired genomic machine learning. F1000Research 2018, 7:233 (doi: 10.12688/f1000research.14048.1)
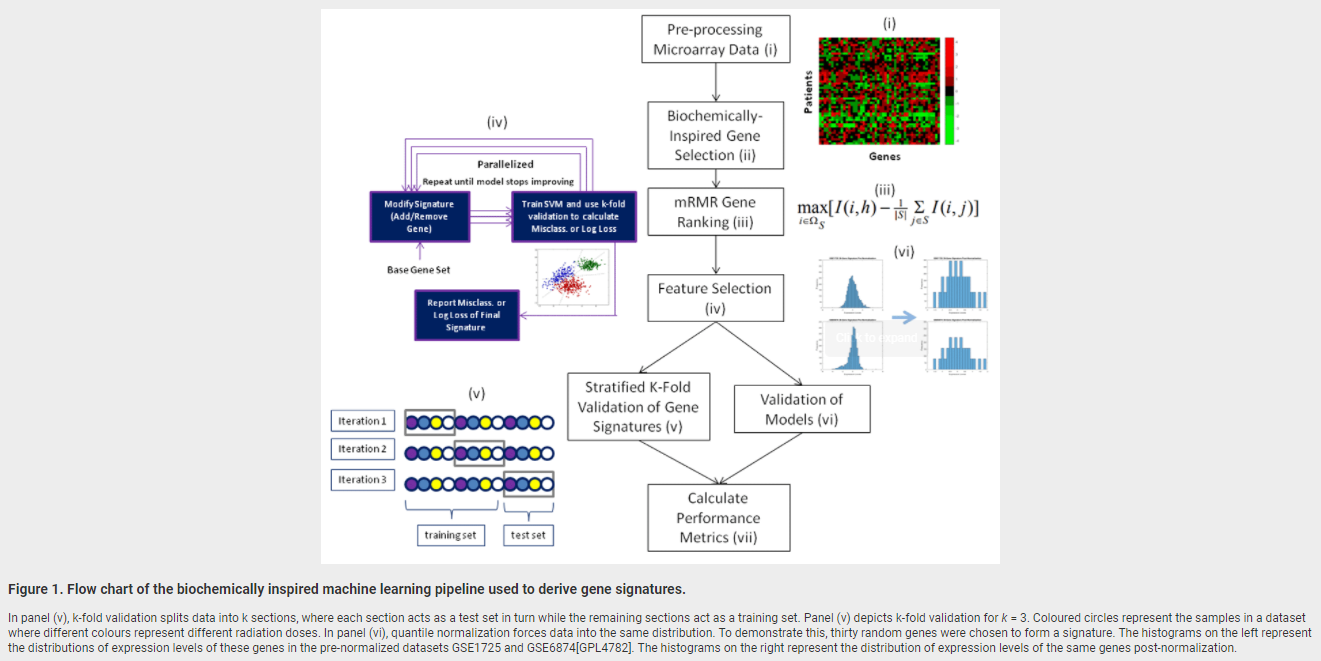
February 19, 2018. Article on genomic signature of radiation exposure
Manuscript describing accurate genomic signatures of radiation exposure will be published shortly by F1000Research.
Jonathan ZL Zhao, Eliseos J Mucaki, Peter K Rogan. Predicting Exposure to Ionizing Radiation by Biochemically-Inspired Genomic Machine Learning, F1000Research, in press.
Abstract:
Background: Gene signatures derived from transcriptomic data using machine learning methods have shown promise for biodosimetry testing. These signatures may not be sufficiently robust for large scale testing, as their performance has not been adequately validated on external, independent datasets. The present study develops human and murine signatures with biochemically-inspired machine learning that are strictly validated using k-fold and traditional approaches.
Methods: Gene Expression Omnibus (GEO) datasets of exposed human and murine lymphocytes were preprocessed via nearest neighbor imputation and expression of genes implicated in the literature to be responsive to radiation exposure (n=998) were then ranked by Minimum Redundancy Maximum Relevance (mRMR). Optimal signatures were derived by backward, complete, and forward sequential feature selection using Support Vector Machines (SVM), and validated using k-fold or traditional validation on independent datasets.
Results: The best human signatures we derived exhibit k-fold validation accuracies of up to 98% (DDB2, PRKDC, TPP2, PTPRE, and GADD45A) when validated over 209 samples and traditional validation accuracies of up to 92% (DDB2, CD8A, TALDO1, PCNA, EIF4G2, LCN2, CDKN1A, PRKCH, ENO1, and PPM1D) when validated over 85 samples. Some human signatures are specific enough to differentiate between chemotherapy and radiotherapy. Certain multi-class murine signatures have sufficient granularity in dose estimation to inform eligibility for cytokine therapy (assuming these signatures could be translated to humans). We compiled a list of the most frequently appearing genes in the top 20 human and mouse signatures. More frequently appearing genes among an ensemble of signatures may indicate greater impact of these genes on the performance within individual signatures. Several genes in the signatures we derived are present in previously proposed signatures.
Conclusions: Gene signatures for ionizing radiation exposure derived by machine learning have low error rates in externally validated, independent datasets, and exhibit high specificity and granularity for dose estimation.
February 7, 2018. Accepted presentations at EPR Biodose (Munich, June, 2018)
Ali, S, Li Y, Shirley B, Wilkins R, Flegal F, Rogan PK, Knoll JHM. Population scale biodosimetry with the Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) software system. [Platform]
Rogan PK, Zhao JZL, and Mucaki EJ. Predicting exposure to ionizing radiation by biochemically-inspired genomic machine learning.[Poster]
Li Y, Shirley B, Wilkins R, Flegal, F, Knoll JHM, Rogan PK. Optimization of image selection in Automated Dicentric Chromosome Analysis. [Poster]

May 4, 2015. Comment on PMID 23348723. Prediction of mutant mRNA splice isoforms by information theory-based exon definition.
Peter Rogan 2015 May 04 6:14 p.m.
The Logic and Formulation of Exon Definition for Splice and Splicing Regulatory Sites with Negative Information Content. PK Rogan, EJ Mucaki
Update on: Mucaki EJ, 2013 and the Automated Splice Site and Exon Definition Analysis server (ASSEDA).
In Mucaki EJ, 2013, we described a method of predicting the overall strength of an exon by calculating its total information content (Ri,total) from the sum of the Ri values of its donor and acceptor splice sites, adjusted for their gap surprisal (the self-information of the distance between the two sites). Differences between ΔRi,total values are predictive of the relative abundance of these exons in distinct processed mRNAs.
Splice sites altered by mutations that prevent stable interaction with splicesomes are said to be abolished. Information theory predicts abolition of binding below their minimum binding affinity, Ri,minimum, which is empirically derived. This value is slightly above zero bits, the theoretical minimum for binding at equilibrium (ΔG = 0; Schneider TD, 1997). Sites with Ri < 0 are not bound, forming stable interactions would be endergonic (ΔG > 0). This raises the question, when predicting the change in exon strength (ΔRi,total) due to a mutation that inactivates binding, whether mutant sites with varying degrees of negative information content are energetically distinguishable from one another.
The computation of Ri,total contains the sum of the the Ri values of component binding sites, irrespective of their initial or final strengths. Thus, a mutated site with Ri << 0 would result in greater ΔRi,total compared to a site with Ri ~ 0. To assess whether the degree of unfavorable binding should be applied to the exon definition calculation, or if values below 0 bits should be computed similarly to a binding site at equilibrium (Ri ~ 0), we reevaluated experimentally validated natural and regulatory splicing mutations in our paper with both approaches. Ri,total was calculated for 10 variants from Supplementary Table 2, both including and excluding the negative information (ie. Ri < 0 vs. Ri = 0) of inactivated splice sites. Mutation #2 of Supplementary Table 2 [ADA:g.43249658G>A] abolishes a natural donor site, from 8.8 to -9.9 bits. In applying the full decrease in strength (ΔRi,total: -18.7 bits), the natural exon strength decreases from 21.0 to 2.3 bits. When the negative information content is set to zero bits, the change is significantly smaller (21.0 -> 12.2 bits; ΔRi,total = -8.8 bits). When a weak natural splice site is abolished, the difference as expressed as ΔRi,total can be quite small (Mutation #9; -14.8 vs -3.1 bits). In the case of Mutation #38, the reduction in ΔRi,total leads to a partially discordant prediction where the abolished natural exon is weaker than the experimentally confirmed activated cryptic exon. Results for this mutation were concordant with the published version when the negative bit value of the mutated natural site was included in the calculation.
The impact of mutations in splicing regulatory (SR) factors can also be predicted on ASSEDA, where the Ri of the SR binding site is added to the R_i,total, as well as a secondary gap surprisal value for the particular SR protein. These sites can also be abolished. But when a SR protein binding site is no longer active, should the SR gap surprisal still be applied, or is the SR gap surprisal no longer applicable?
We tested mutations from Mucaki EJ, 2013 (Supplementary Table 4), which abolish the splicing enhancer SF2/ASF with and without the SR protein gap surprisal when Ri of the SR site is < 0 bits. The removal of the gap surprisal term for Mutation #2 of Supplementary Table 4 leads to a discordant prediction, where the ΔRi is less than the SR gap surprisal at that distance and therefore the ΔRi,total is positive. As experimental evidence shows an increase in skipping, it is a discordant prediction. Therefore, the gap surprisal is still applied in the computation of both initial and final Ri,total values when the SR protein of interest is abolished as the site is naturally present and therefore expected for binding. Conversely, when we apply the gap surprisal to the initial Ri,total for a splicing factor that is being created, we are essentially applying a penalty for a site that does not normally exist. Therefore, we no longer apply the SR gap surprisal value to the initial Ri,total in these cases.
The revised Ri,total values of SR binding site mutations slightly differ from those reported in Mucaki EJ, 2013 (Supplementary Table 4). This is because the gap surprisal distributions were recomputed for the following factors: SF2/ASF, SC35 and SRp40, with updated versions of these models based on CLIP Seq data (Blin K, 2015, Khorshid M, 2011). This resulted in small changes to the distributions for SF2/ASF and SC35, however changes for SRp40 were significant, and now more closely resembles the other gap surprisal functions. The updated graphs of distance vs. gap surprisal are available at: http://splice.uwo.ca/gapsurprisals.html. While this should not significantly affect ΔRi,total values, it may affect the initial and final Ri,total values.
-
This article was mentioned in a comment by Peter Rogan2018 Jan 12 2:39 p.m.
-
This article was mentioned in a comment by Peter Rogan2017 Dec 12 09:53 a.m.
See:Analysis of the effects of rare variants on splicing identifies alterations in GABAA receptor genes in autism spectrum disorder individuals.[Eur J Hum Genet. 2013.]
-
This article was mentioned in a comment by Peter Rogan2017 Dec 07 5:24 p.m.
See:Characterization of a novel germline BRCA1 splice variant, c.5332+4delA. [Breast Cancer Res Treat. 2017.]
-
This article was mentioned in a comment by Peter Rogan2017 Oct 01 8:57 p.m.
See:Rules and tools to predict the splicing effects of exonic and intronic mutations. [Wiley Interdiscip Rev RNA. 2018.]