Peter Rogan will be presenting “A Unified Framework For Prioritization Of Variants Of Uncertain Significance In Hereditary Breast And Ovarian Cancer” at Variant Detection 2017 in Santiago de Compostela Spain on June 5, 2017.
Coauthors are Eliseos Mucaki1, Natasha Caminsky1, Ruipeng Lu1, Joan Knoll1,2 and Peter Rogan1,2. 1University of Western Ontario, 2CytoGnomix Inc.
Abstract:
Purpose: A significant proportion of HBOC patients receive uninformative genetic testing results, an issue exacerbated by the overwhelming quantity of variants of uncertain significance identified. We apply information theory (IT) to predict and analyze non-coding variants of uncertain significance (VUS) in regulatory, coding, and intronic regions based on changes in binding sites in these genes. This provides a unifying framework where, aside from protein coding changes, pathogenic variants occurring within sequence elements can be prioritized based 19 on their recognition by proteins involved in mRNA splicing, transcription, and untranslated region binding and structure. To support the utilization of IT analysis, we established IT-based variant interpretation accuracy by performing a comprehensive review of mutations altering mRNA splicing in rare and common diseases1.
Methods: We captured and enriched for coding and non-coding variants in genes known or suspected to increase HBOC risk. Custom oligonucleotide baits spanning the complete coding, non-coding, and intergenic regions 10 kb up- and downstream of ATM, BRCA1, BRCA2, CDH1, CHEK2, PALB2, TP53, ATM, BARD1, BRCA1, BRCA2, CDH1, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51B, STK11, TP53, and XRCC2 were synthesized for solution hybridization enrichment. Aside from protein coding and copy number changes, IT-based sequence analysis was used to identify and prioritize pathogenic non-coding variants that occurred within sequence elements predicted to be recognized by proteins or protein complexes involved in mRNA splicing, transcription, and untranslated region (UTR) binding and structure. Mutation-associated affinity changes were computed in transcription factor (TFBSs)2, splicing regulatory (SRBSs)3, and RNA-binding protein (RBBSs)4 binding sites following mutation. This approach was supplemented by in silico and laboratory analysis of UTR structure.
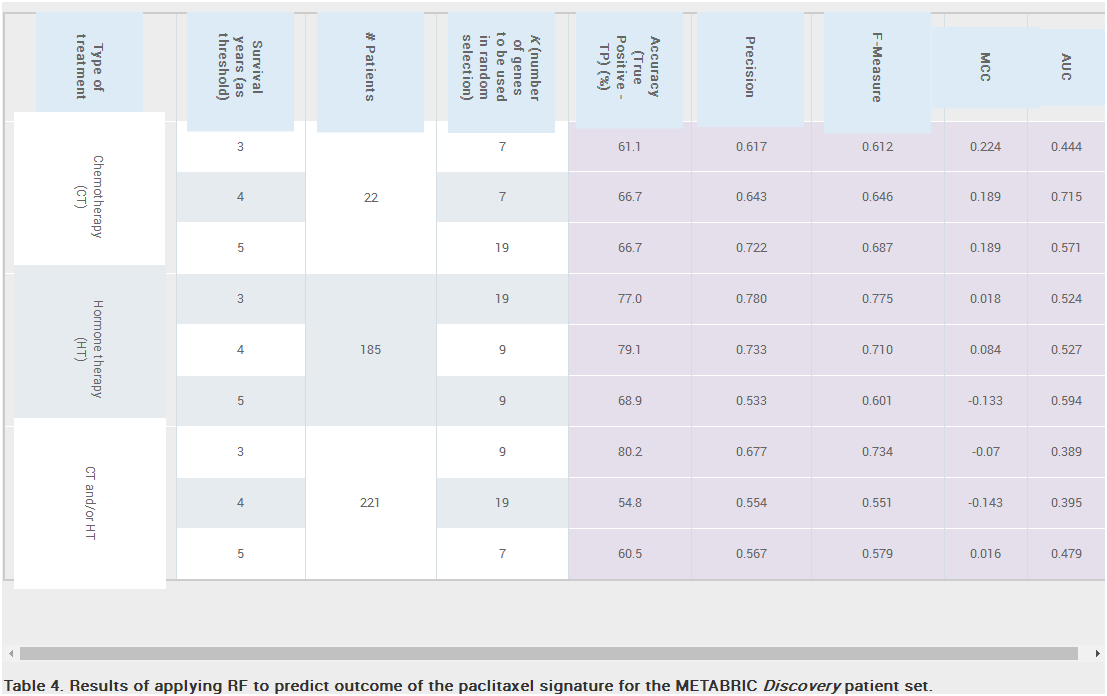
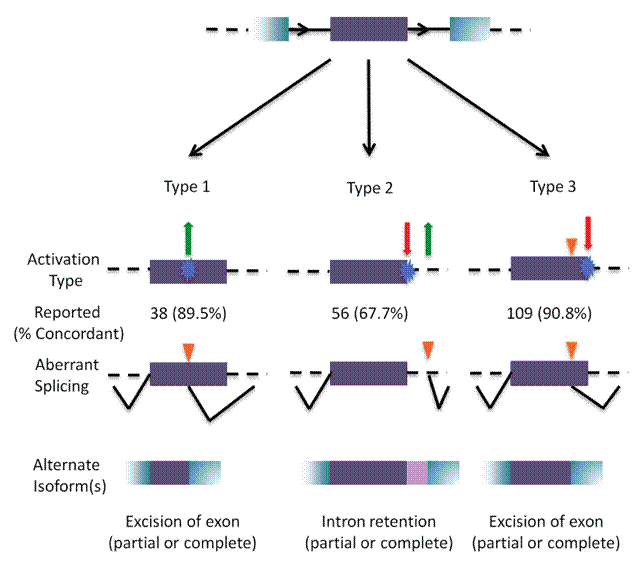
Results: Unique and divergent repetitive sequences were sequenced in 379 high-risk, patients without identified mutations in BRCA1/2. We identified 47,501 unique variants and we prioritized 429 variants. The methods were first applied in 7 complete genes (ATM, BRCA1, BRCA2, CDH1, CHEK2, PALB2, TP53) in 102 anonymized individuals (15,311 variants)4, then validated in 287 patients in an ethics board approved study (38,372 variants)5. In the validation study, we prioritized variants affecting the strengths of 10 splice sites (4 natural, 6 cryptic), 148 SRBS, 36 TFBS, and 31 RBBS. Three variants were also prioritized based on their predicted effects on mRNA secondary (2°) structure, and 17 for pseudoexon activation. Additionally, 4 frameshift, 2 in-frame deletions, and 5 stopgain mutations were identified. Multifactorial cosegregation analysis further reduced the set of candidate pathogenic variants in some families.
Conclusion: Complete gene sequence analysis followed by a unified framework can be used to interpret non-coding variants that may affect gene expression. When combined with pedigree information, complete gene sequence analysis can distill large numbers of VUS among a wide spectrum of functional mutation types to a limited set of variants for downstream functional and co-segregation analysis. References: 1Caminsky et al. F1000Res 3:282, 2015; 2Lu et al. Nucleic Acids Res. doi: 10.1093/nar/gkw1036, 2016; 3Mucaki et al. Hum. Mut. 34:557-565. 2013; 4Mucaki et al. BMC Med Genomics. 9:19, 2016; 5Caminsky et al. Hum. Mut. 37:640-52, 2016.